

| Journal of Clinical Gynecology and Obstetrics, ISSN 1927-1271 print, 1927-128X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Gynecol Obstet and Elmer Press Inc |
| Journal website http://www.jcgo.org |
Original Article
Volume 5, Number 4, December 2016, pages 112-116
Outcome of Fetuses With Abnormal Cavum Septi Pellucidi: Experience of a Tertiary Center
George Lucian Zorilaa, Stefania Tudorachea, Elena-Madalina Barbua, Maria-Cristina Comanescua, Razvan-Grigoras Capitanescua, Marius-Cristian Marinasb, Maria Floreaa, Nicolae Cerneaa, Dominic Gabriel Iliescua
aPrenatal Diagnosis Unit, Department of Obstetrics and Gynecology, University Emergency County Hospital, Craiova, Romania
bDepartment of Anatomy, University of Medicine and Pharmacy of Craiova, Romania
cCorresponding Author: Stefania Tudorache, Prenatal Diagnosis Unit, Department of Obstetrics and Gynecology, University Emergency County Hospital, Craiova, Romania
Manuscript accepted for publication December 28, 2016
Short title: Fetuses With Abnormal Cavum Septi Pellucidi
doi: https://doi.org/10.14740/jcgo423w
| Abstract | ▴Top |
Background: Cavum septi pellucidi (CSP) is easily evaluated in the second and third trimester of the pregnancy. The structure is an important feature of the standard planes used for routine morphological assessment of fetal head and central nervous system (CNS): trans-thalamic and trans-ventricular plane. The standard description of the CSP is an anechoic rectangular box between two hyperechoic lines represented by the septum pellucidum. The pathological aspects are mainly represented by the absence of its normal appearance, which is associated with severe CNS malformations of the brain midline as corpus callosum agenesis, hydranencephaly, porencephaly, schizencephaly, holoprosencephaly, syntelencephaly or severe chronic hydrocephaly. Other issues such as increased or reduced dimensions of the CSP are not considered significant if found isolated, although the fetal genetic investigation is suggested by some authors. The objective of the study was to evaluate the outcome of fetuses with abnormal CSP detected by sonography in the last 5 years, in the Prenatal Diagnosis Unit (PDU) of our tertiary center.
Methods: We performed a retrospective review of the cases with abnormal CSP evaluated in our tertiary unit, diagnosed between January 2012 and November 2016. The fetal anatomy was evaluated in all cases following the recommendations of the international guidelines. In abnormal CSP cases, fetal neurosonogram and an extended fetal anomaly scan were performed and amniocentesis was proposed to identify genetic disorders.
Results: A total of 7,520 cases were examined for morphological purposes and abnormal CSP was found in 36 cases. Absent CSP was the initial observation that triggered further investigation and diagnosis in the cases with agenesis of corpus callosum (ACC) (seven cases) and septo-optic dysplasia (two cases). In hydranencephaly or severe hydrocephaly, porencephaly, schizencephaly and holoprosencephaly, the heavily malformed aspect of the brain is obvious, and the absence of CSP is only an observation, with less diagnostic importance and clinical implications. Partial or total ACC was mainly associated to absent CSP, as the development of two structures is merged. Almost half of the total abnormal CSP cases (16/36) were associated with genetic disorders, most of them with abnormal karyotype and all of them were associated with the absence of CSP. The persistent enlargement of CSP (4/36 cases) and the hyperechoic aspect of CSP (1/36) were not associated with other structural or genetic abnormalities and the postnatal neuromotor development was normal.
Conclusions: The CSP evaluation is mandatory and normal aspects suggest a normal development of the midbrain. Genetic testing should be offered especially for the cases with absent CSP because of the high incidence of chromosomal disorders. As the absence of CSP is associated with severe structural or genetic disorders, its visualization in the second half of pregnancy is mandatory for any anomaly scan. The enlargement and echogenicity variations of CSP associate a favorable neonatal outcome. However, long-term follow-up is recommended in the apparently normal neonates and infants, as they may develop abnormal psychological behavior lately.
Keywords: Cavum septi pellucidi; Corpus callosum; Ultrasound scan; Neurosonogram; Prenatal diagnosis; Central nervous system
| Introduction | ▴Top |
Cavum septi pellucidi (CSP) is an important structure, developed together with the corpus callosum (CC) starting from 10 weeks gestation age (GA) until 18 weeks GA. It is located in the middle of the brain, placed above the fornix, between the two medial walls of the lateral ventricles and below the CC. This structure is continued posteriorly with cavum vergae and the limit between these two is a plane between the Monroe foramens. Most of the authors use the term CSP for “CSP and Vergae”. We comply with this term and use “CSP” for both of the structures. The septi pellucidi serves as an important relay station; its most important anatomical and functional fiber connections are with the hippocampus and the hypothalamus.
CSP is routinely imaged after 18 gestational weeks on the three mandatory views of the fetal head obtained during fetal sonography. Specifically, in addition to the images of the ventricles and the posterior fossa, an axial view at the level of the paired thalami yields both the biparietal diameter and the CSP [1-8]. An important issue for the abnormal CSP diagnostic is not to mistake fornix for CSP, because of their proximity and relative similar appearance. The normal CSP should appear as a dark box due to the fluid within the cavum, surrounded by two white lines on both lateral sides, represented by the septum pellucidum. At the level of the fornix, the box is divided by a third sagittal line. The differentiation is of major importance, as the fornix development is not related to CC [9].
Abnormal aspects of CSP include its absence, enlargement or echogenicity. The normal widening of CSP has been standardized between 2 and 4.7 mm (± 2 SD) at 19 - 20 weeks GA and up to 9 mm as an upper limit at 38 weeks, considering 2 SD as well [10]. The enlargement of the CSP was associated with hydrocephaly, chromosomal translocations and growth restriction. When found as an isolated sonographic abnormality, the fetal outcome is favorable, although fetal genetic evaluation should be offered [11-13]. The normal evolution of CSP after birth is closure from cavum vergae to CSP, and only 15% of this space is visible at 6 months after birth. An enlarged CSP of more than 1 cm after birth, and the persistence of a CSP after infancy have been described as “subtle markers of cerebral dysgenesis”, possibly associated with neuropsychiatric disturbances, particularly schizophrenia. Thus, postpartum follow-up of cases with prenatal enlargement of CSP is important [14].
The absence of CSP in the second and third trimester has been associated with ACC, septo-optic dysplasia, hydranencephaly, porencephaly, schizencephaly, holoprosencephaly, syntelencephaly or severe chronic hydrocephaly [1-10, 14].
The prenatal population large studies regarding the morpho-genetic associations of abnormal CSP and their rates are useful for the prenatal parental counseling. Given the low prevalence of the anomaly, and its heterogeneity, a large number of cases are needed. Our series is one of the largest in literature, and the spectrum of our data offers the possibility of inclusion in larger meta-analysis. Our objective was to report the outcome of the cases with any abnormal aspect of CSP that does not comply with the normalcy criteria, e.g. abnormal echogenicity.
| Methods | ▴Top |
The prenatal characteristics were retrospectively analyzed from the ultrasound database, over a 5-year period, between January 2012 and November 2016. The cases were examined in a tertiary center - the Prenatal Diagnosis Unit (PDU) of the University Emergency County Hospital, Craiova, Romania. The fetal anatomy was evaluated in all cases following the recommendations of the international guidelines [8]. The examinations were performed by obstetricians with special interest in fetal medicine, with minimum 2 years of experience in prenatal US. The cases with suspected abnormalities that could not be confirmed during the initial examination were invited for reassessment. Whenever abnormalities were suspected at US examination, two experienced examiners studied the fetus to confirm the anomalies. In abnormal CSP cases, fetal neurosonogram and an extended fetal anomaly scan were performed and amniocentesis was proposed to identify genetic disorders. 2D and 3D fetal ultrasounds were carried out in all cases with CNS abnormalities (Voluson 730 Pro US machines, GE Medical Systems). Post-processing of the acquired volumes was intended for a better visualization of the abnormalities or the associated features. After confirmation, appropriate counseling and management were provided by interdisciplinary team (obstetrician, genetician, neonatologist - pediatrician, pediatric surgeon).
| Results | ▴Top |
During the time interval, we examined 7,520 pregnancies for morphologic purposes, CNS malformations were found in 257 cases, and the main anomaly was ventriculomegaly (Fig. 1). Abnormal CSP was found in 36 cases as follows: the absence of CSP was found in 31 cases (86.1% from CSP anomalies), an enlarged CSP was measured in four cases (11.1%) and hyperechoic CSP was noted in one case (2.7%).
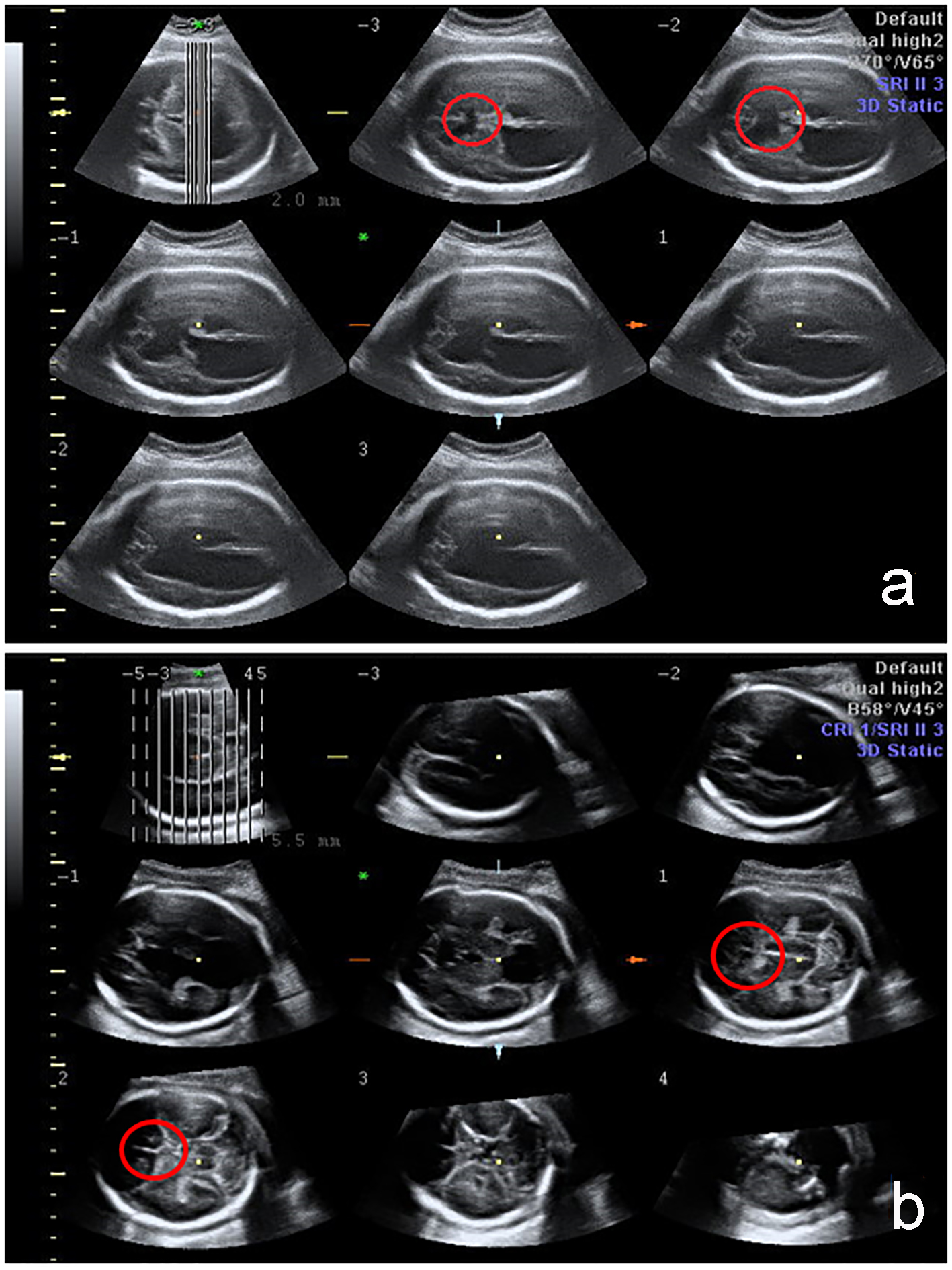 Click for large image | Figure 1. 3D fetal CNS evaluation with tomographic ultrasound imaging (TUI). The absence of CSP is indicated by the red circle in cases with severe brain anomalies: holoprosencephaly and schizencephaly. |
The CNS abnormalities (Fig. 2) associated with the absence of CSP were: agenesis of corpus callosum in seven cases (22.5%), holoprosencephaly (HPE) in three cases (9.6%), syntelencephaly in two cases (6.4%), porencephaly in two cases (6.4%), massive hydrocephaly in five cases (16.1%), anencephaly in four cases (12.9%), schizencephaly in two cases (6.4%), encephalocele in one case (3.2%) and septo-optic dysplasia in three cases (9.6%). Dandy-Walker malformation was associated to ACC on three of the seven cases.
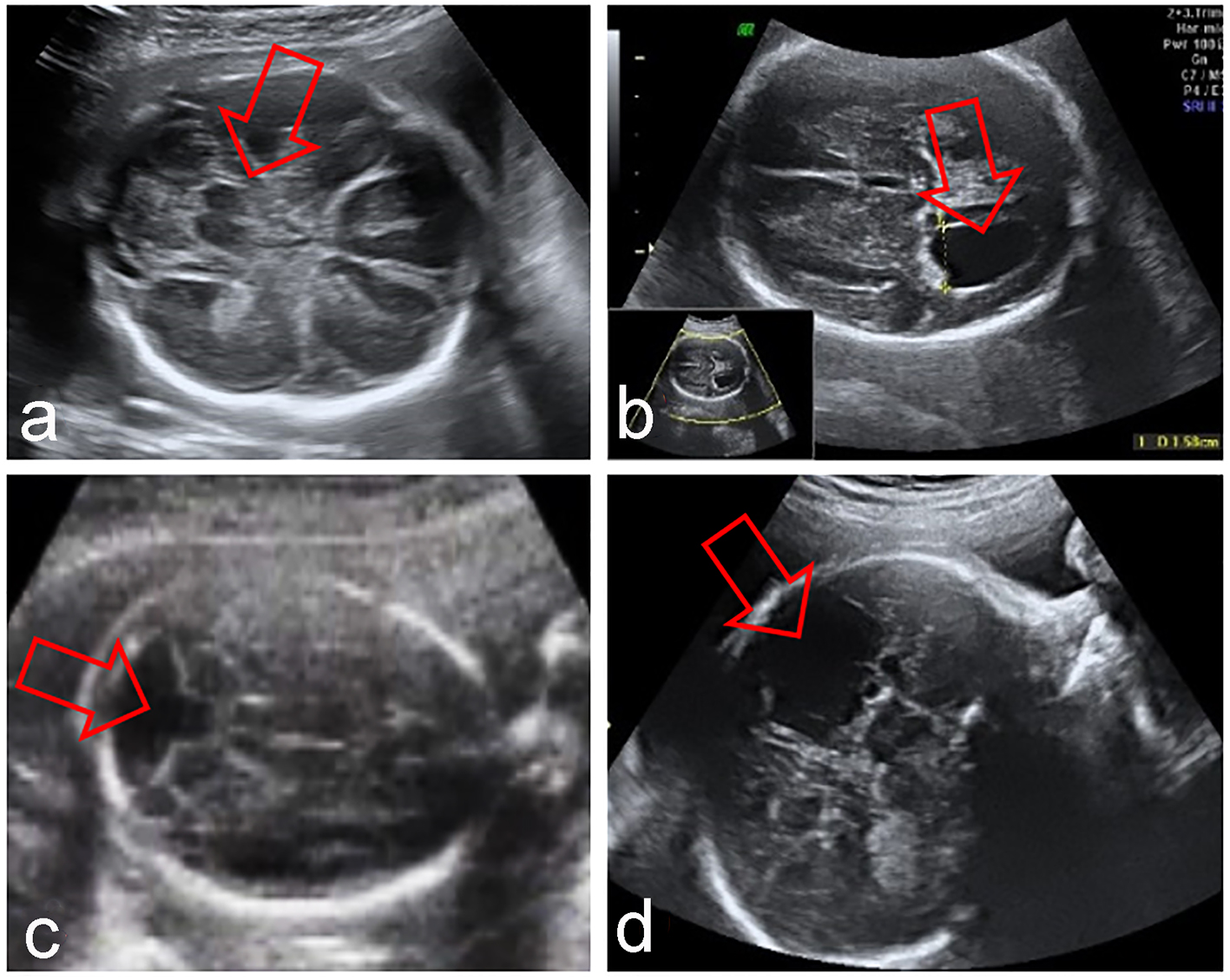 Click for large image | Figure 2. Fetal CNS abnormalities associated to absent CSP: syntelencephaly (a), hydrocephaly (b), Dandy-Walker syndrome (c), porencephaly (d). |
In two cases, CSP was absent in pregnancies after 37 GW (6.4%), at their admission to our unit, as a first antenatal imagistic evaluation.
We considered one case of hyperechoic CSP as “abnormal”, as it does not fulfill normal CSP criteria: “a black box between two white lines”. Persistent enlargement of CSP (Fig. 3), if consider two standard deviations as cut-off limit, was found in four cases. All of them had no other structural anomaly, and the genetic tests yielded normal results.
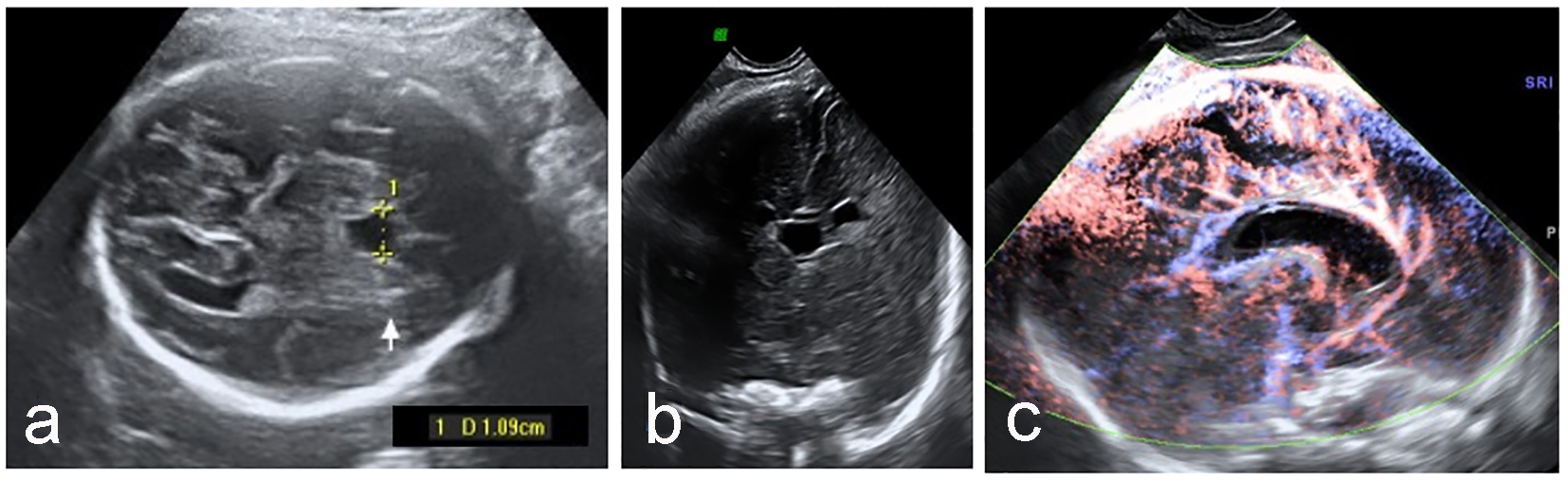 Click for large image | Figure 3. Enlarged CSP (a) with normal anterior cerebral complex in coronal plane (b) and pericallosal artery (c). |
Genetic tests were available in 24 CSP abnormal cases, and disorders were found in 16 cases, all associated with absent CSP (Table 1). Genetic disorders were found in 16 cases, half of the cases with absence of CSP. Most frequent genetic disorders were trisomies (21,18,13), but we also found other chromosomal alteration, deletions and translocations.
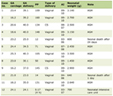 Click to view | Table 1. Spectrum of CNS Abnormalities and the Associated Abnormalities |
| Discussion | ▴Top |
A normal CSP suggests a normal development of the prozencephalon and may rule out some commonly found CNS structural anomalies. Three ultrasound studies, including a total of 1,024 normal fetuses, during the second and third trimester, showed that the CSP can be visualized and measured in 100% of normal fetuses between 20 and 37 weeks of gestation [13, 15, 16].
The CSP closure starts from posterior cavum vaergae to anterior real space of CSP. It has been communicated that cavum is present at term in 85% of the cases and in 15% at 6 months after birth [14]. We found two cases of absent CSP in ultrasound examinations performed beyond 37 weeks. This cases did not present associated anomalies. Their presentation for fetal ultrasound was the first during the current pregnancy.
The CSP development is strongly related to corpus callosum, and most of the authors concluded that normal CSP equals normal CC. Although non-visualization of the CSP is not synonymous with agenesis of the corpus callosum, these two entities are frequently associated. As corpus callosum is not routinely visualized during the basic CNS evaluation at the fetal anomaly scan, the identification of a normal appearence of CSP is of major importance in the detection of ACC. Still, the CSP may not be visualized in spite of normal CC [1-10, 14]. As the absence of CSP is mostly associated to ACC, in such cases the first step is to evaluate the development of CC in axial, coronal and sagital views and to visualize the pericalosal artery using Doppler investigation of the sagittal plane. MRI and tractography are useful to confirm the ultrasound aspects [17, 18]. In our study, the absence of CSP was associated with ACC in seven cases (two cases as partial and five as total agenesis). In four of these cases (57%), non-cerebral structural anomalies were described and other CNS anomalies were found in about 42% of all cases. We found two out of seven cases with Dandy-Walker syndrome - most of the authors reported approximately one-third of cases [14, 17] and in one case spina bifida was present.
Another major abnormality that benefits from CSP visualization is septo-optic dysplasia. In this condition, the only prenatal ultrasound abnormal sign may be the absence of CSP, therefore the routine CSP evaluation is very important for the detection of these cases, although MRI is crucial for diagnosis confirmation. There were two Morsier syndrome cases (septo-optic dysplasia) that associated absence of CSP with normal CC. CSP was absent and a slight communication between the lateral ventricular anterior horns was present, while the CC was normally developed. The condition was confirmed postpartum and one of these cases associated unilateral labial cleft. In the other case, right aortic arch with left ductus arteriosus was found.
There were 24/36 genetic exams available (cytogenetic and molecular samples). Disorders have been found in 16 abnormal CSP cases, from which 13 were abnormal karyotype and the other three were abnormal molecular array CGH results. Genetic assessment is mandatory especially for absence of CSP, karyotype and microarray being also highly recommended [19].
Absent CSP may be associated to a large number of structural disorders as holoprosencephaly (HPE), from alobar to lobar, and the more recently described entity of syntelencephaly; septo-optic dysplasia (SOD); callosal dysgenesis and hypogenesis; chronic severe hydrocephalus, typically from aqueduct stenosis or the Chiari II malformation; schizencephaly; porencephaly, hydranencephaly; basilar encephaloceles; and isolated septal deficiency [1-10, 13, 14-17, 19]. In hydranencephaly, porencephaly, schizencephaly, holoprosencephaly and severe hydrocephaly, the heavily malstructured aspect of the brain is obvious, and the abnormality of CSP is only an observation, but with less importance in the diagnostic and prognostic of the cases.
The real clinical importance of an enlarged prenatal CSP is unknown, but it has been stressed that this finding should be followed by a detailed search for associated anomalies and postnatal imaging and developmental evaluation is indicated. The persistence of a wide CSP after birth has been related to schizophrenia if its wideness exceeds 10 mm, thus a close follow-up is required in these cases [11-14].
Hyperechoic CSP has not been described in literature up to date, therefore a close follow-up of our isolated case is quite important. The prenatal extended investigations of this case as MRI and genetic tests yielded normal results. The infant had a good progress in the first year.
Conclusions
Absent CSP is a very important CNS malformation marker, and its presence should trigger extended fetal morpho-genetic evaluation. In our experience, almost half of the cases associated genetic disorders and 39% associated structural malformations.
Other aspects as isolated enlargement or hyperechoic CSP need further evaluation in larger studies on a long-term outcome in order to establish their significance, as our cases presented a normal postpartum initial evolution, but the number of these abnormalities communicated in the literature is low.
| References | ▴Top |
- AIUM Practice Guideline for the performance of an antepartum obstetric ultrasound examination. J Ultrasound Med. 2003;22(10):1116-1125.
pubmed - AIUM practice guideline for the performance of obstetric ultrasound examinations. J Ultrasound Med. 2010;29(1):157-166.
pubmed - Filly RA, Cardoza JD, Goldstein RB, Barkovich AJ. Detection of fetal central nervous system anomalies: a practical level of effort for a routine sonogram. Radiology. 1989;172(2):403-408.
doi pubmed - Nyberg DA. Recommendations for obstetric sonography in the evaluation of the fetal cranium. Radiology. 1989;172(2):309-311.
doi pubmed - Gushiken BJ, Goldstein RB. Practical approach to evaluating the fetal neural axis. Semin Roentgenol. 1999;34(1):5-12.
doi - ACOG Practice Bulletin No. 58. Ultrasonography in pregnancy. Obstet Gynecol. 2004;104(6):1449-1458.
doi - Angtuaco TL. Ultrasound imaging of fetal brain abnormalities: three essential anatomical levels. Ultrasound Q. 2005;21(4):287-294.
doi pubmed - Sonographic examination of the fetal central nervous system: guidelines for performing the 'basic examination' and the 'fetal neurosonogram'. Ultrasound Obstet Gynecol. 2007;29(1):109-116.
doi pubmed - Callen PW, Callen AL, Glenn OA, Toi A. Columns of the fornix, not to be mistaken for the cavum septi pellucidi on prenatal sonography. J Ultrasound Med. 2008;27(1):25-31.
pubmed - Timor-Tritsch E. Ultrasonography of the prenatal brain, third edition. McGraw Hill, 2012; 2:62-65, 3:128, 6:235-244.
- Sherer DM, Sokolovski M, Dalloul M, Santoso P, Curcio J, Abulafia O. Prenatal diagnosis of dilated cavum septum pellucidum et vergae. Am J Perinatol. 2004;21(5):247-251.
doi pubmed - Bronshtein M, Weiner Z. Prenatal diagnosis of dilated cava septi pellucidi et vergae: associated anomalies, differential diagnosis, and pregnancy outcome. Obstet Gynecol. 1992;80(5):838-842.
pubmed - Jou HJ, Shyu MK, Wu SC, Chen SM, Su CH, Hsieh FJ. Ultrasound measurement of the fetal cavum septi pellucidi. Ultrasound Obstet Gynecol. 1998;12(6):419-421.
doi pubmed - Winter TC, Kennedy AM, Byrne J, Woodward PJ. The cavum septi pellucidi: why is it important? J Ultrasound Med. 2010;29(3):427-444.
pubmed - Falco P, Gabrielli S, Visentin A, Perolo A, Pilu G, Bovicelli L. Transabdominal sonography of the cavum septum pellucidum in normal fetuses in the second and third trimesters of pregnancy. Ultrasound Obstet Gynecol. 2000;16(6):549-553.
doi pubmed - Serhatlioglu S, Kocakoc E, Kiris A, Sapmaz E, Boztosun Y, Bozgeyik Z. Sonographic measurement of the fetal cerebellum, cisterna magna, and cavum septum pellucidum in normal fetuses in the second and third trimesters of pregnancy. J Clin Ultrasound. 2003;31(4):194-200.
doi pubmed - Paladini D, Pastore G, Cavallaro A, Massaro M, Nappi C. Agenesis of the fetal corpus callosum: sonographic signs change with advancing gestational age. Ultrasound Obstet Gynecol. 2013;42(6):687-690.
doi pubmed - Mitter C, Prayer D, Brugger PC, Weber M, Kasprian G. In vivo tractography of fetal association fibers. PLoS One. 2015;10(3):e0119536.
doi pubmed - Sarwar M. The septum pellucidum: normal and abnormal. AJNR Am J Neuroradiol. 1989;10(5):989-1005.
pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Gynecology and Obstetrics is published by Elmer Press Inc.