

| Journal of Clinical Gynecology and Obstetrics, ISSN 1927-1271 print, 1927-128X online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Clin Gynecol Obstet and Elmer Press Inc |
| Journal website https://www.jcgo.org |
Case Report
Volume 10, Number 2, June 2021, pages 51-54

Successful Management of Significant Maternal 3 Beta-Hydroxysteroid Dehydrogenase Deficiency
Samantha Edwardsa, d, Amy Marinoa, c, Dana Phillipsb, Jennifer Phyb
aTexas Tech University Health Sciences Center, School of Medicine, 3601 4th Street Lubbock, TX 79430, USA
bDepartment of OB-GYN, Texas Tech University Health Sciences Center, 3601 4th Street Lubbock, TX 79430 USA
cU.S. Anesthesia Partners, 6606 Lyndon B Johnson Fwy, Suite 200 Dallas, TX 75240, USA
dCorresponding Author: Samantha Edwards, Texas Tech University Health Sciences Center, School of Medicine, 3601 4th Street Lubbock, TX 79430, USA
Manuscript submitted February 19, 2021, accepted May 11, 2021, published online June 24, 2021
Short title: Management of Maternal 3β-HSD Deficiency
doi: https://doi.org/10.14740/jcgo730
| Abstract | ▴Top |
We report a case of successful management of pregnancy in a patient with significant 3β-hydroxysteroid dehydrogenase (3β-HSD) deficiency. A 25-year-old female presented for interfertility evaluation after failure to conceive for 8 months. The patient began full menses each month at the time of ovulation. Her unique clinical presentation led to the diagnosis of a significant 3β-HSD deficiency. Treatment for infertility with clomiphene citrate, gonadotropins, dexamethasone and human chorionic gonadotropin trigger was unsuccessful. The patient conceived spontaneously while taking continuous combination oral contraceptive pills. Due to her history of instability and full menses in the luteal phase, oral estrogen and progesterone in oil were administered to support the early pregnancy. The results of elevated 17-hydroxyprogesterone (17-OHP), dehydroepiandrosterone (DHEA), and ratio of 17 OH-pregnenolone to 17-OHP were consistent with diagnosis of significant 3β-HSD deficiency. Successful pregnancy was achieved after exogenous hormone supplementation. Fertility in 3β-HSD deficiency, a rare form of congenital adrenal hyperplasia, has not been extensively studied. Varying phenotype can make the diagnosis challenging. Heightened suspicion in patients with reported bleeding at ovulation or breakthrough bleeding on oral contraceptive pills could aid in the diagnosis. Successful pregnancy in these patients is possible.
Keywords: 3β-HSD deficiency; Congenital adrenal hyperplasia; 17-OH-pregnenolone; Periovulatory bleeding; Luteal phase defect
| Introduction | ▴Top |
Congenital adrenal hyperplasia (CAH) is associated with deficiencies of 21-hydroxylase, 11 β-hydroxylase and 3β-hydroxysteroid dehydrogenase (3β-HSD) [1]. The most prevalent of these defects, 21-hydroxylase deficiency, affects only adrenal hormone production and produces hypertension and virilization. A deficiency of 3β-HSD type II occurs in approximately 0.5% of all cases of CAH and affects both adrenal and gonadal steroidogenesis [2]. The enzyme 3β-HSD converts pregnenolone to progesterone to feed the pathway that ultimately produces aldosterone [3]. A second action is the conversion of 17-hydroxypregnenolone (17-OH-pregneolone) to 17-hydroxyprogesterone (17-OHP) to ultimately synthesize cortisone in the adrenal cortex. Dehydroepiandrosterone (DHEA) conversion to androstenedione is a third action, which is important for the production of androgens and estrogens in both the adrenals and gonads (Fig. 1). Mutations of 3β-HSD are thought to be the result of nonsense, frameshift, and missense mutations, with an autosomal-recessive mode of inheritance [4]. The enzyme exists in two isomeric forms: type I is expressed in the placenta and peripheral tissues; and type II is expressed in the adrenal gland, ovary, and testes [1]. Type II is the isoenzyme most commonly mutated in 3β-HSD deficiency and results in gonadal dysfunction (oligomenorrhea) in women [4]. The severity of the genetic defect results in a variety of phenotypes. A deficiency of 3β-HSD classically presents with some degree of salt wasting, mild virilization, or symptoms suggestive of polycystic ovary syndrome (PCOS) [4, 5]. Non-classical cases of 3β-HSD deficiency can lack developmental abnormalities because DHEA acts as a weak androgen, therefore, in moderate amounts may not induce virilization. This subset of patients may remain undiagnosed until presenting later in life with acne, hirsutism, and menstrual irregularities. Patients with a 3β-HSD defect are commonly infertile because of menstrual irregularities, including oligo-ovulation and primary amenorrhea [2]. In a study conducted by Schram et al, 60% of the study participants with 3β-HSD deficiency presented with menstrual irregularities [6]. A detailed literature review revealed limited data related to fertility and chance of successful pregnancy in patients with 3β-HSD deficiency.
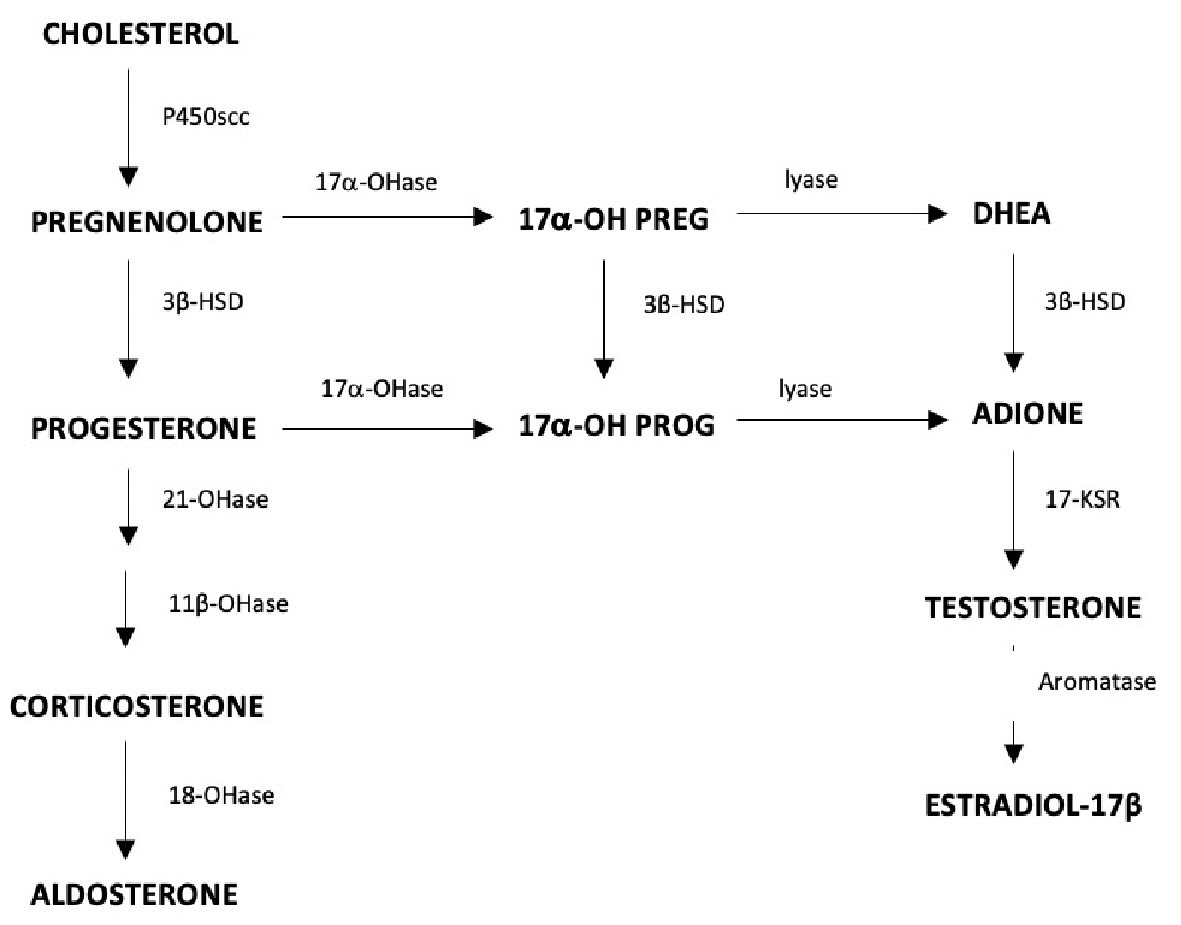 Click for large image | Figure 1. Ovarian steroidogenesis, the role of 3β-HSD in the conversion of DHEA to androgens and estrogens in the adrenal glands and gonads. 3β-HSD: 3β-hydroxysteroid dehydrogenase; DHEA: dehydroepiandrosterone. |
| Case Report | ▴Top |
Investigations
We present a case of a 25-year-old female who was initially referred to a reproductive endocrinology clinic at age of 25 with infertility, endometriosis and irregular menstrual cycles after 8 months of unsuccessful conception. Since menarche at age 15, her cycle pattern was 14 days of mild to heavy spotting followed by 10 days without bleeding. The patient’s vital signs were blood pressure 80/60 mm Hg, pulse 64/min, respirations 12/min, height 175 cm, weight 57.6 kg, body mass index (BMI) 18.8 kg/m2, and temperature 36.5 °C. The patient was lean and had mild hirsutism on physical exam.
Diagnosis
Initial laboratory tests revealed estradiol, progesterone, follicle-stimulating hormone (FSH), luteinizing hormone (LH), prolactin, thyroid-stimulating hormone (TSH), testosterone and electrolytes were all within reference ranges. The patient had elevated 17-OHP which prompted adrenocorticotropic hormone (ACTH) stimulation testing to check for CAH. Cortrosyn® (250 mg) was administered intravenous (IV) during the patient’s follicular phase and results demonstrated elevated 17-OHP, DHEA, and ratio of 17-OH-pregnenolone to 17-OHP, indicating a significant 3β-HSD deficiency (Table 1) [7, 8]. Transvaginal ultrasound revealed polycystic appearing ovaries.
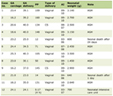 Click to view | Table 1. Results of ACTH (Cortrosyn®) Stimulation Test |
Treatment
We attempted daily dexamethasone (2.5 mg) therapy for 2 months followed by combination therapy with clomiphene citrate (50 mg/day) and additional gonadotropin resulted in follicular development. However, as the patient reached day of human chorionic gonadotropin (hCG) trigger, she began to have spotting and eventually full menses in the luteal phase. The patient underwent four such attempted treatments without success. She then adopted two children.
After cessation of fertility treatment, the patient desired treatment for her symptomatic endometriosis and was treated with leuprolide acetate (3.75 mg intramuscularly) for 4 months, followed by a monophasic extended-cycle oral contraceptive (OCP, 30 µg ethinyl estradiol and 150 µg levonorgestrel, Seasonale®). Breakthrough bleeding occurred and the patient was started on a triphasic 35 µg ethinyl estradiol/norgestimate OCP (Ortho Tri-Cyclen®), which also resulted in breakthrough bleeding. Similar episodes of breakthrough bleeding occurred with subsequent 20 µg ethinyl estradiol and 0.3 mg drospirenone combination OCP (Yaz®). An extended-cycle OCP (0.03 mg ethinyl estradiol and 0.15 mg levonorgestrel for 84 days with 0.01 mg ethinyl estradiol for 7 days, Seasonique®) was prescribed.
Follow-up and outcomes
The patient spontaneously conceived while taking these pills continuously. Pregnancy was confirmed by quantitative BhCG level at 3 and 4/7 weeks. The patient stated that she took a pregnancy test because it was unusual for her not to have bleeding at that time on the continuous OCPs. Review of the literature again showed no previous successful pregnancy in women with significant 3 β-HSD deficiency. Given the patient’s history of long-term significant luteal phase defect, we administered oral estradiol and intramuscular progesterone therapy comparable to our standard frozen embryo transfer protocol to the patient after she consented to it. Oral estrogen and intramuscular progesterone were continued until 12 weeks gestation. The patient delivered a healthy female infant at term.
| Discussion | ▴Top |
3β-HSD deficiency is a rare cause of CAH, with a highly variable clinical presentation [1, 2, 9]. Literature review revealed that little is known about fertility in women with 3β-HSD deficiency, as the hypothalamic-pituitary-gonadal axis is variable. Subtle signs such as irregular menses, faint hirsutism and polycystic ovary-like changes are observed in some non-salt wasting females with 3β-HSD deficiency [10]. Our patient had a partial block of 3β-HSD, resulting in increased levels of 17-OHP and pregnenolone. Elevated levels of 17-OHP suggests CAH, because low cortisol production increases ACTH release, resulting in increased circulating levels of 17-OHP [11]. To distinguish the type of CAH in cases of increased 17-OHP, an ACTH stimulation test is recommended. After ACTH stimulation, if levels of 17-OH-pregnenolone exceed 100 nmol/L and the ratio of 17-OHP to 17-OH progesterone is high, a defect in 3β-HSD is suspected [7]. For non-classical cases of 3β-HSD deficiency, dexamethasone suppression is typically recommended to promote menstrual regularity in addition to clomiphene citrate to increase follicle production [12]. Schram et al concluded that eight out of nine patients studied reported improvement with irregular menses after 3 months of glucocorticoid use. Additionally, there was reported improvement in acne and some cases of hirsutism [6]. Improved fertility has been reported, but results are equivocal. The patient presented in this case failed to conceive after 4 months of daily dexamethasone therapy followed by combination therapy with clomiphene citrate and gonadotropins.
Elevated levels of progestins, in this case 17-OHP, inhibit follicular growth and endometrial proliferation [12]. Fertility may be further compromised by thickened cervical mucus and involution of the endometrium. Shram et al reported one patient who did not show improvement of menstrual irregularities after 2 years of glucocorticoid therapy. However, menses became regular after cyclical use of estrogen and progesterone [6]. Although the mechanism leading to a successful conception in this case is not known, we suggest that the spontaneous conception may have occurred after supplying exogenous hormones at levels sufficient to induce ovulation. 3β-HSD is an enzyme that predominantly occurs in the gonads. 3β-HSD deficiency results in decreased production of all three groups of adrenal steroids including glucocorticoids, mineralocorticoids and sex steroids. Many women with classic or non-classic forms of CAH remain anovulatory with conventional fertility treatments most likely due to abnormally elevated progesterone levels in the follicular phase, or in the case of our patient, elevated pregnenolone. Combined OCP pills likely suppressed pregnenolone and down-stream androgen production enough to increase gonadotropin-releasing hormone pulse frequency allowing follicular development and spontaneous ovulation. Further study of partial 3β-HSD deficiencies could provide more insight into the mechanism of conception while taking continuous OCPs. To our knowledge, this is the first case reporting successful pregnancy and live birth in a patient with significant 3β-HSD deficiency. We hypothesize that after providing sufficient luteal support, normal maintenance of pregnancy can be achieved because of the production of functional placental type I 3β-HSD [1].
Learning points
This report illustrates a unique case of 3β-HSD deficiency, a rare form of CAH that presented with subtle symptomology, varied responses to hormonal stimulation and suppression, and ultimately resulted in a spontaneous pregnancy during continuous combination OCPs. Increased clinical suspicion for a partial 3β-HSD deficiency should accompany patients with significant periovulatory bleeding or breakthrough bleeding while compliant on OCPs. While successful pregnancy in patients with 3β-HSD deficiency is possible, more research is needed to better understand and recognize this rare form of CAH and to facilitate successful conception and management of pregnancy.
Acknowledgments
The authors would like to thank our patient for her consent in presenting her case.
Financial Disclosure
No fund, grant, or other support was received.
Conflict of Interest
The authors have no conflict of interest to declare that is relevant to the content of this article.
Informed Consent
The patient’s informed consent for publication of this report was obtained.
Author Contributions
All authors contributed to the conception, design, writing and critical editing of the manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Adashi EY, Hennebold JD. Single-gene mutations resulting in reproductive dysfunction in women. N Engl J Med. 1999;340(9):709-718.
doi pubmed - Al Alawi AM, Nordenstrom A, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase type 2 deficiency. Endocrine. 2019;63(3):407-421.
doi pubmed - Pang S, Wang W, Rich B, David R, Chang YT, Carbunaru G, Myers SE, et al. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2002;87(6):2556-2563.
doi pubmed - Garner PR. Congenital adrenal hyperplasia in pregnancy. Semin Perinatol. 1998;22(6):446-456.
doi - New MI. Diagnosis and management of congenital adrenal hyperplasia. Annu Rev Med. 1998;49:311-328.
doi pubmed - Schram P, Zerah M, Mani P, Jewelewicz R, Jaffe S, New MI. Nonclassical 3 beta-hydroxysteroid dehydrogenase deficiency: a review of our experience with 25 female patients. Fertil Steril. 1992;58(1):129-136.
doi - Simard J, Moisan AM, Morel Y. Congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase/Delta(5)-Delta(4) isomerase deficiency. Semin Reprod Med. 2002;20(3):255-276.
doi pubmed - Lutfallah C, Wang W, Mason JI, Chang YT, Haider A, Rich B, Castro-Magana M, et al. Newly proposed hormonal criteria via genotypic proof for type II 3beta-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87(6):2611-2622.
doi pubmed - Rojansky N, Shushan A, Rosler A, Weinstein D, Laufer N. Long-term infertility in late-onset 3 beta-ol-dehydrogenase deficiency: successful pregnancy following dexamethasone and in vitro fertilization (IVF) therapy. J In Vitro Fert Embryo Transf. 1991;8(5):298-300.
doi pubmed - Pang S. Congenital adrenal hyperplasia owing to 3 beta-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am. 2001;30(1):81-99, vi-vii.
doi - Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, Han TS, et al. Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. 2010;95(11):5110-5121.
doi pubmed - Stikkelbroeck NM, Hermus AR, Braat DD, Otten BJ. Fertility in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Obstet Gynecol Surv. 2003;58(4):275-284.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Clinical Gynecology and Obstetrics is published by Elmer Press Inc.